Neuropathology And Neurochemistry
The pathological hallmark of PD was considered to consist of intracellular eosinophilic inclusions called Lewy bodies. These occur inside neurons in the substantia nigra, presumably in dopamine (DA)-containing cells. Lewy bodies probably accumulate in neurons undergoing degeneration. The number of DA neurons in the substantia nigra progressively diminishes in PD. It is important to note that only DA neurons in the substantia nigra whose axons are destined to go to the putamen (less so to the caudate) in the nigrostriatal tract are affected, while other dopaminergic tracts are spared, with progressive loss of DA in the striatum. Clinical symptoms first appear when DA content in the striatum is reduced by about 70%. This may imply that a long preclinical stage, of 20 years or more, predates the appearance of motor symptoms. Other neurotransmitter systems are also affected in PD. These include norepinephrine (NE) loss in the cell bodies of the locus coeruleus, serotonin (5-hydroxytryptamine, 5-HT) loss in the raphe nuclei, and cholinergic cell loss in the nucleus basalis of Meynert. These deficiencies probably contribute to the affective and cognitive changes in PD but may also contribute to motor dysfunction. However, it is clear that most motor disturbances are primarily related to DA deficits, because replacement of endogenous DA can dramatically alleviate the motor disability. Until recently, it was impossible to demonstrate the DA deficiencies during life. However, this was changed by the use of positron emission tomography (PET) and single photon emission tomography (SPECT). Using radioactive tracer techniques, it can be demonstrated that DA marker accumulation is reduced in the corpus striatum in PD, presumably because of the loss of DA terminals that normally take up these markers. PET and SPECT correlate with the side of the clinical abnormalities in unilateral parkinsonism, and may be sensitive enough to detect the progression of the disease (Bhatt et al., 1991). While the complete chemical composition of Lewy bodies is not yet known, they definitely contain parkin, ubiquitin, and synuclein. Antibodies against these substances, particularly anti-synuclein antibodies, will stain all Lewy bodies in the substantia nigra and frequently additional ones in the brain stem, olfactory bulb, and in the cortex. Anti-synuclein staining in the substantia nigra relates to motor dysfunction. Braak et al. (2004) have suggested a sequential progression of the pathology in PD, starting in the dorsal motor nucleus of the vagus and the olfactory bulb, than appearing in other brainstem nuclei and the substantia nigra, and only at a late stage in the cerebral cortex. Interestingly, staining in neurites is also seen. These Lewy neurites (Braak et al., 1996) may suggest axonal dysfunction as an important part of the pathophysiology of PD.Pathophysiology
The pathophysiology of PD is still rather clouded. The dopaminergic denervation of the basal ganglia (and particularly the striatum) is obviously central in the movement abnormalities. ‘Motor loops’ involving the basal ganglia, subthalamic nucleus, thalamus, and cortex have been described (Bergman et al., 1990). However, their function is poorly understood. How the loss of dopamine causes tremor at rest, enhanced tone both at rest and during action, and bradykinesia or hypokinesia still needs to be fully explained. The pathophysiology of the fourth cardinal feature of PD, loss of postural reflexes, is even less clear. Some gains were made through the use of kinematic studies, such as of arm trajectories. These quantify the defects and demonstrate some unexpected findings (e.g., regarding the importance of visual feedback), explaining how the ‘motor loops’ incorporate sensory information (Flash et al., 1991). The nigrostriatal pathway activates, in the corpus striatum, dopaminergic receptors. Five subtypes have been identified and, while the most important seem to be of the D2 type, the role of D1 receptors in normal brain activity is unclear, and therefore the functional correlate of activation of these receptors is not established. In particular, it is not clear whether activation of D1 receptors is important for the elicitation of dyskinesias or other motor complications occurring commonly in advanced stages of the disease in patients who are treated by levodopa.Pathogenesis Of PD
There is no consensus on the pathogenesis of the disease. The fact that only a selected population of neurons die off may suggest the involvement of a toxin affecting these cells. Drugs are known that can selectively damage catecholaminergic neurons – for example, 6-hydroxydopamine (6-OHDA). This substance is uptaken by the DA transporters, and it is concentrated in DA cells and causes their degeneration. Because 6-OHDA does not cross the blood–brain barrier (BBB), it cannot account for human PD. (However, it is an important experimental tool in the study of PD and drug development.) But another chemical, MPTP, has been identified as causing in humans a disorder quite similar to PD in many characteristics. The mechanisms of MPTP toxicity have been explored in depth and, although there is no doubt that this chemical only accounts for very few cases of PD, the possible existence of MPTP-like chemicals has been explored. Epidemiological and toxicological studies have inconsistently suggested an environmental toxin (Stevenson et al., 1989). The possibility of endogenous production of a substance similar to MPTP in its mechanism of action is still debated (Tanner and Langston, 1990). The substantia nigra and globus pallidum are rich in iron; the iron concentration increases with age and particularly in PD. This may suggest involvement of this metal in neurotoxicity, perhaps through a process of lipid peroxidation.Genetics
About 10% of patients with PD report first-degree relatives with the same disorder. Although they constitute only a small portion, these cases are important in many ways. During the past two decades, several genes have been identified that account for monogenic forms. In addition to being able to help in genetic counseling, these genes have been very important in understanding the mechanism underlying the neurodegenerative processes in PD. For example, the first gene to be described, a-synuclein, could have either point mutation or overexpression (duplication or triplication). The protein synuclein was found to be a major component of the Lewy bodies. It is likely that this protein, once mutated or overexpressed, will be more likely to be misfolded, thus evading the usual mechanisms of intracellular protein metabolism. In attempt to be removed from the cell, it is directed into the Lewy bodies. Another gene that was found to be associated with PD is parkin, coding for an enzyme responsible for intracellular degradation of proteins (such as a-synuclein). Another gene, PINK1, codes for a mitochondrial enzyme (mitochondrial dysfunction has long been thought to be a cause of PD). The most common genetic abnormality underlying PD is the LRRK2 gene, which may account for 1–2% of PD cases and in some ethnic groups much more frequently. The genetic heterogeneity associated with PD is helpful in discovering mechanisms of neurodegeneration. But even more surprising is the finding of pathologic heterogeneity: Patients carrying the parkin mutations usually do not have Lewy bodies, while in families with LRRK2 mutations, some members may contain Lewy bodies in nigral neurons while others do not. These observations shatter the view that PD is a Lewy body disease, as if patients without Lewy bodies must have a ‘different disease’ to explain their parkinsonism. Several other mutations have been described (Table 1) (Polymeropoulos et al., 1997; Gasser et al., 1998; Kitada et al., 1998; Kruger et al., 1998; Leroy et al., 1998; Bonifati et al., 2002; Hicks et al., 2002; Singleton et al., 2003; Pankratz et al., 2003; Paisan-Ruiz et al., 2004; Valente et al., 2004; Zimprich et al., 2004; Ramirez et al., 2006).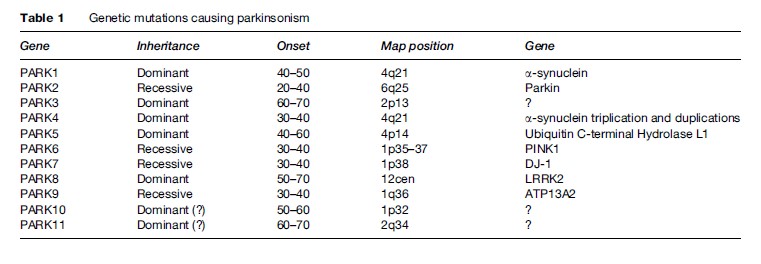
Treatment Of PD
The basic treatment of PD is by replacement of the deficient DA using levodopa. Levodopa is absorbed from the gastrointestinal tract and transported through the blood–brain barrier by active amino acid transport mechanisms. In the brain, as well as in the periphery, levodopa is metabolized to DA by an enzyme, 1-amino acid decarboxylase. This enzyme can be blocked by the substances benserazide and carbidopa. Employing either of these inhibitors can prevent the peripheral conversion of levodopa to DA, thus diverting larger amounts of levodopa to the brain, where it is taken up by dopaminergic terminals to be used in dopamine synthesis. Most patients today are treated by a combination of levodopa and one of those enzyme inhibitors. By preventing the peripheral conversion to DA, undesirable side effects such as orthostatic hypotension and nausea are minimized. A second enzyme involved in levodopa metabolism is catechol O-methyltransferase (COMT). The inactivation of DA after its release into the synaptic cleft involves both reuptake by DA terminals and metabolism. COMT inhibitors, tolcapone and entacapone, are used clinically together with levodopa because they prevent peripheral metabolism of levodopa. By prolonging the half-life of levodopa, they minimize fluctuation of serum concentration which may cause subsequent adverse events. The reuptake is performed by specialized DA transporter molecules in the membrane. Inhibitors of this transporter, as well as those of COMT, may prolong the action of DA. Levodopa replacement is extremely effective in controlling much of the disability in PD. It is most efficacious against rigidity and hypokinesia, but tremor also responds. However, postural instability does not respond well to dopaminergic therapy. Because the progressive loss of DA neurons continues despite levodopa treatment, patients become less and less mobile as time elapses since the last dose was ingested, manifesting as end-of-dose hypokinesia. Therefore patients gradually require higher doses of the drug. These increments may cause significant problems, particularly peak-dose dyskinesias. Basically, these reactions are to be expected because when brain DA concentrations are very high, the patient is in a state opposite to the baseline DA deficiency.Treating patients who reach this stage can be done by dividing the daily dose into several smaller administrations. While initially three daily doses of levodopa are sufficient to control symptoms, as the disease advances six or more doses may be required. In normal subjects, levodopa will never produce dyskinesias. Presumably this is because terminals of the nigrostriatal pathways in the corpus striatum take up any excessive DA and either store or degrade it to inactive metabolites. This buffering mechanism will necessarily fail in PD because of the progressive loss of DA neurons and terminals (Karstaedt and Pincus, 1992). The loss of this buffering capacity may be responsible also for the eventual and most problematic complication of therapy, the so-called ‘on-off ’ phenomenon. Patients fluctuate from being normal in their function, or even dyskinetic as a manifestation of excessive DA stimulation (‘on’), to severe parkinsonian hypokinesia and rigidity (‘off ’). As the disease advances, these fluctuations come on unexpectedly (‘random on-off ’). Once buffering capacity is lost, pharmacokinetic factors (e.g., levodopa serum concentration) determine the clinical response state (Korczyn, 1973). Motor fluctuations could be due to erratic absorption of levodopa from the gastrointestinal tract (possibly related partly to competition by amino acids derived from dietary proteins), distribution factors, or transportation across the blood–brain barrier. Attempts to reduce such fluctuations, which are of some benefit, include a low-protein diet (Karstaedt and Pincus, 1992), gastric administration of levodopa at a constant rate or by duodenal infusion (Antonini et al., 2007; Samanta and Hauser, 2007), controlled-release levodopa preparations, and administration of direct-acting dopamine agonists (DAA) (Rascol et al., 2000), either orally or, in advanced stages, as watersoluble DAA (e.g., rotigotine, lisuride, and apomorphine) (Parkinson Study Group, 2003; Pahwa et al., 2006; Korczyn, 2007; Poewe et al., 2007). The revolutionary introduction of levodopa into the therapeutics of PD was so dramatic that its impact is unlikely to be superseded by another drug any time soon. However, as is discussed above, this treatment does not solve all the problems. One critical question relates to the time at which levodopa therapy should be initiated. The basic aim of levodopa therapy is to replace endogenous DA. It is thus a symptomatic therapy that, however, also masks to some extent the relentless progression of neuronal cell loss. However, it is still unclear whether levodopa treatment itself accelerates or retards this loss. There are suggestions that levodopa reduces the oxidative stress that results from excessive burden on the remaining neurons. Alternatively, it is possible that the pharmacological concentrations of extrinsic levodopa will contribute to the formation of toxic free radicals inside neurons. Therefore, diverging views exist on whether levodopa should be started immediately upon diagnosing PD, or delayed as much as possible, with the aid of other types of therapy (Rascol et al., 2002). Monoamine oxidase (MAO), the enzyme that metabolizes several catecholamines and indolamines, exists in two forms. MAO-A metabolizes not only dopamine but also NE and 5-HT, whereas MAO-B does not metabolize either NE or 5-HT. Selective inhibitors of MAO-B, and particularly selegiline (deprenyl) and rasagiline, are effective against MPTP toxicity. In PD patients, selegiline and rasagiline provide symptomatic benefit (Olanow and Calne, 1992). This may be related to an ampfetaminelike action in releasing DA from terminals or, more likely, by preventing DA reuptake. Interestingly, it has been shown that newly diagnosed PD patients can be maintained on MAO-B inhibitors alone for a long period (Landau, 1990; Olanow and Calne, 1992), although the significance of this observation is still unclear (Bonucelli and Del Dotto, 2006). Monotherapy with selegiline or rasagiline is not efficacious in more advanced cases. At present, many patients are being treated with selegiline or rasagiline in addition to levodopa. The usefulness of this combination in retarding the progression of the disease has not been convincingly demonstrated, although rasagiline is useful in moderating the motor fluctuations in advanced stages of PD (Bonucelli and Del Dotto, 2006). Direct-acting dopamine agonists (DAA) are important in the treatment of PD. These include apomorphine, bromocriptine, pergolide, cabergoline, and lisuride, as well as newer agents such as rotigotine, ropinirole, and pramipexole. Theoretically, the use of such agents could be advantageous. In initial stages, they relieve the excessive burden on remaining DA neurons without being subject to metabolism into toxic free radicals inside DA neurons, as has been hypothesized for levodopa. In later stages, it is easier to maintain constant levels at receptor sites because these drugs do not depend on active transport in the gut and through the BBB. Particularly cabergoline, which has a very long biological half-life, may be advantageous in PD patients who develop motor fluctuations or off symptoms at night (Inzelberg et al., 1995). Similar benefit may occur if percutaneous administration of DA agonists is applied, such as rotigotine and lisuride. However, DAA have significant drawbacks and side effects. Their potency is lower than that of levodopa. Therefore they can be used as monotherapy in the initial stages of the disease but will have to be supplanted by levodopa in subsequent years. Ergoline derivatives are not very specific and interact with several subtypes of DA receptors as well as with 5-HT and other receptors. D1 stimulation may contribute to the occurrence of dyskinesias, while 5-HT and D4 stimulation may be conductive to hallucinations and other psychiatric manifestations. Ropinirole and pramipexole, two synthetic nonergoline compounds, are specific to DA (particularly D2 type) receptors and were therefore expected to be advantageous. Unfortunately, this does not seem to be the case, and the frequency of these psychiatric adverse events is identical. In addition, DA agonists act also at the periphery, and this may contribute to significant side effects such as orthostatic hypotension and nausea and leg edema (Rascol et al., 2007). A behavioral syndrome consisting of gambling, uncontrolled shopping or eating, and hypersexuality has been reported and may be rather severe (Weintraub et al., 2006). These behavioral effects were termed dopamine dysregulation syndrome and are probably related to the effect of dopamine agonists on D3 and D4 receptors, since in animals quetiapine, a dopamine agonist that activates these receptors selectively, has been shown to induce stereotypic ‘compulsive’ behavior. Also, recently cardiotoxic effects were described for two agents, cabergoline and pergolide, leading to the withdrawal of pergolide from the market (Roth, 2007). Surgical interventions of PD are also available. These include ablative and transplanting approaches. Targets for functional stereotactic neurosurgical lesions that reduce tremor are the ventrolateral thalamus and the posteroventral pallidum. Within the last decade, accumulating evidence has proved that subthalamic stimulation is very effective in the treatment of PD, reducing both the parkinsonian symptoms and dyskinesias, and allowing reduction of drug dosage (Sailer et al., 2007). Whether subthalamic stimulation should be initiated at an early stage of the disease is still an open question. There has been extensive interest in transplanting DA tissue into the caudate or putamen in PD. Originally, autologous tissue was used, but the benefits, if any, were offset by the significant complications (Windner and Rechcronal, 1993). This approach was discarded. In newer experiments, dopaminergic transplants were used in which the tissue was removed from aborted fetal midbrains. The use of stem cells is presently being explored (Sontag et al., 2005). It is difficult to assess the success of this approach, because frequently the patients who have been recruited had a poor prognosis to start with and also because this intervention is associated with a high placebo factor (Korczyn, 1993). However, the main question may not necessarily be whether neurons are generated, survive, and produce and release dopamine, but whether the release of dopamine can be properly regulated, since otherwise on-off dyskinesias will result.Cognitive Changes In PD
The prevalence of frank dementia in PD is far greater than that in the general population. PD dementia may be preceded by mild memory loss, transient confusional episodes, vivid dreams, or hallucinosis. Clinically, the dementia of PD differs from that of Alzheimer disease (AD). PD patients rarely develop dysfunctions of the isocortical association areas, such as dysphasia or agnosia, and may resemble a ‘frontal’ type of dementia, with dysexecutive symptoms. But while the differentiation between cortical and subcortical dementia is of some theoretical interest, individual PD patients may develop a clinical AD-like picture. Cell loss in PD is not limited to DA neurons. The degeneration of cholinergic neurons in the nucleus basalis of Meynert, as well as 5-HT, NE, and somatostatin deficiencies are well-documented, and glutamatergic deficiency may also exist. These deficiencies are similar to those observed in AD and therefore suggest similarities in pathogenesis and treatment, as well as a clinical overlap. During the past decade, it has become obvious that Lewy bodies are not limited to the substantia nigra in PD, but may occur in a widespread distribution, extending to the cortex. Diffuse Lewy body disease is a pathological entity whose clinical correlates have been tentatively defined (McKeith et al., 2005). Patients commonly have cognitive decline and parkinsonian features, and either one may dominate the picture. Therefore, it seems that Lewy body disease can first manifest itself as parkinsonism (if the lesions primarily affect the substantia nigra) or cognitive decline (if predominantly the cortex is affected). There seems to be a continuum in this respect, and the question is whether factors can be identified that are responsible for which region is more affected. The main risk factors for dementia in PD include older age and severity of motor symptoms (Giladi et al., 2000). Treatment of the cognitive changes of PD is unsatisfactory. The cholinergic defect suggests that drugs with antimuscarinic action may be detrimental, and these include not only specific antiparkinsonian agents such as benzhexol or trihexyphenidyl but also antidepressants such as amitriptyline. Contrariwise, cholinomimetic agents such as rivastigmine, widely used in AD, may be of significant value in dementia associated with PD (Giladi et al., 2003; Emre et al., 2004). Treatment of hallucinations and delusions similarly poses difficult problems because the use of D2 blockers may well result in motor exacerbation. Clozapine, a specific D4 blocker, has been suggested as an efficacious treatment of this condition (Rabey et al., 1995).Depression In PD
Exactly how frequently depression occurs in PD is a question that is difficult to answer. There is quite a spectrum of figures in the literature, which diverge depending on (1) the criteria used to diagnose depression, (2) possible inclusion or exclusion of demented patients or those with parkinsonism due to causes other than PD (e.g., vascular etiology and progressive supranuclear palsy), and (3) the severity of the neurological impairment. In addition, referral bias to specialized centers probably results in excessive numbers of depressed patients in these centers. However, and regardless of these factors, it is safe to conclude that depression is rather common in PD. Because depression is potentially treatable, this conclusion is of significant importance.

Our Advantages
- Quality Work
- Unlimited Revisions
- Affordable Pricing
- 24/7 Support
- Fast Delivery
Order Now